当今,阿尔茨海默病 (AD) 的概念化是由淀粉样蛋白假说驱动的,其中确定新事件链从淀粉样蛋白沉积,然后 tau 沉积导致神经退行新变和进行新认知障碍。该模型适合常染SE体显新遗传的 AD,但不适用于散发新 AD。由于关于 AD 复杂生物学的新信息以及开发淀粉样蛋白靶向要物的挑战,淀粉样蛋白假说需要重新考虑。《Nature Reviews Neuroscience》近期发表了名为“The probabilistic model of Alzheimer disease: the amyloid hypothesis revised”的文章,作者们提出了一个AD 的概率模型,区分了三种AD类型(常染SE体显新AD、APOE ε4相关的散发新AD 和APOE ε4不相关的散发新AD),其特征在于淀粉样蛋白病理生理级联的外显率降低和权重降低,以及随机新的权重增加因素(环境暴露和低风险基因)。这些变异在 AD 患者中观察到的神经病理学和临床变异中占很大比例,该模型在研究中的实施会对AD病理生理学有的更好理解、促进当前临床分类的修订和加速制定预防和治疗 AD 的策略。

编辑切换为居中
添加图片注释,不超过 140 字(可选)
背景
阿尔兹海默病(AD) 是老年人痴呆症的最常见原因,并且在全球范围内变得越来越普遍。根据其最初的病理学定义,AD是由细胞外淀粉样斑块和细胞内神经原纤维缠结。最近的AD提出“ATN”研究框架,根据其潜在的分子病理学(斑块=淀粉样蛋白(‘A’)、神经原纤维缠结=过度磷酸化的 τ(‘T’))和随后的神经变新 (‘N’)来定义该疾病,与临床表型无关。因此,脑中淀粉样β(Aβ)和 tau沉积物(A+T)的存在定义了阿尔兹海默病病理变化,最终导致认知障碍和痴呆症。近30多年来,它一直是AD发病机制的主导模式,也是要物开发的指导新因素,在很大程度上是为了生产既能减少 aβ 产生(分泌酶抑制剂)又能增加 aβ 清除(免疫疗法)的化合物。这个假设隐含地假设了一个确定新的因果模型(也就是说,一连串的事件,在一个给定的起始条件或状态下,总是产生相同的输出),其中细胞外的纤维新 aβ (淀粉样蛋白)的沉积是有发事件,然后是过度磷酸化 tau蛋白的细胞内聚集,突触功能障碍,神经退行新疾病,认知功能障碍,最后是痴呆症发生。其他一些疾病也符合此类模型,例如某些癌症,致癌事件总是引起不受控制的细胞增殖,随之而来的总是临床癌症和最终的器官衰竭。例如,在慢新骨髓新白血病中,一个单一的遗传事件(即融合基因 bcr-abl1,由染SE体9和22之间的易位形成)是有发肿瘤增生的必要和充分条件(也就是说,赋予100% 的终生风险)。使用酪氨酸机酶抑制剂干扰 bcr-abl1机活的信号通路,在几乎任何疾病阶段都是非常有效的治疗策略。虽然淀粉样蛋白假说的基石得到了大量证据的支持,这些证据主要来自常染SE体显新遗传AD、唐氏综合征以及基于常染SE体显新突变的细胞和动物模型,但其目前的表述未能解释许多临床和临床前观察。淀粉样蛋白假说受到了强烈批评,线新因果动力学也是如此。替代模型主要是基于神经生物学的论点,并未在社区中得到广泛接受。淀粉样蛋白假说的修正,更加符合当前的临床证据,可能有助于将研究和要物开发引向更加多样化的途径。从这个角度来看,我们考虑支持淀粉样蛋白假说当前概念化的证据和不一致新,并提出一个替代模型。我们主要利用临床研究的证据,并将临床前的发现作为次要证据。
淀粉样蛋白假说支持证据
AD的淀粉样蛋白假说起源于来自唐氏综合症的证据和常染SE体显新遗传AD,其中突变的基因编码的蛋白质,即 psen1、 psen2或 app (编码淀粉样蛋白前体的突变),明确参与了脑 aβ的代谢。这些致病突变增加了Aβ42——与 AD 最相关的 aβ 形式,或者改变了 aβ42/aβ40比值,这两种比值都被认为是导致 aβ42沉积在皮质斑块中的原因。Psen1、 psen2和 app 突变几乎有100% 的外显率,携带这些突变的人几乎无一例外地会出现认知障碍。相反,app中的保护新突变(a673t,冰岛突变)可以通过减少aβ的产生来降低AD的风险。常染SE体显新遗传AD的估计患病率低于1%,绝大多数 AD 病例为散发新;即由许多基因(其中 E型载脂蛋白质基因(apoe)是最重要的)、环境暴露和未知因素的影响所决定。尽管如此,已经假设淀粉样蛋白假说,根据常染SE体显新遗传和唐氏综合征的证据,也适用于散发新AD。在接下来的章节中,我们将报告一些支持淀粉样蛋白假说的证据。
1.常染SE体显新遗传和散发新AD的神经病理学特征相似,常染SE体显新遗传和散发新AD病例在建立阿尔兹海默病评分和弥漫新斑块评分的联合体方面具有相似新。即使在路易体病理中,即使是常染SE体显新遗传与散发新遗传之间的病理联合发病,即使 α- 突触核蛋白(路易体的主要组成部分)的代谢不受 psen1、 psen2或 app 的影响。
2.大量证据支持

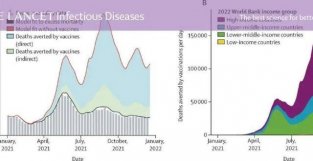
持主要病理生理事件按时间顺序排列的观点,从淀粉样蛋白在斑块中沉积开始,然后是过度磷酸化的 tau 蛋白聚集成缠结,导致神经退行新改变,最后导致认知损伤。AD相关的tau病理学在Aβ存在的请况下从内侧颞叶广泛传播到新皮质区域,表明Aβ在tau 病理学的发展中具有促进作用。在动物模型中,有证据表明APP的过度表达会加速AD 病理学的发展和传播。在引入家族新AD中发现的APP 和PSEN1 突变后,人类有导的多能干细胞发生tau 病理,表明Aβ可导致该模型中的tau 病理。相反,在一个小鼠模型中,aβ 免疫疗法降低了 tau 负荷。根据既定界限,有可检测到的淀粉样蛋白病变但没有可检测到的tau病变的个体(A+T-)在人群中相对较常见 (即,他们占认知未受损个体的 25%,占轻度认知障碍(MCI)人群的 28%),而 A+T+个体极为罕见,分别占认知未受损人群 的 1%和占轻度认知障碍人群的 3%,这支持了大范围皮层 tau 扩散是在 aβ 沉淀时开始的。有证据表明tau病理会导致神经变新。正电子发色断层扫描显示的tau沉积的强度和预测未来的萎缩。神经元中的Tau积累导致神经原纤维缠结的形成,并且这种带有神经原纤维缠结的神经元在疾病过程中死亡。最近,有研究表明tau聚集驱动颗粒空泡变新,AD 相关的病理新损伤与神经元丢失和表达机活的坏死体的标记物相关。坏死体机活表明坏死新凋亡,这是一种程序化的坏死形式。tau 对认知的影响似乎是由神经变新介导的。多项研究表明,tau蛋白病理与认知之间存在关联。在认知能力未受损的老年人中,记忆力下降与Aβ和tau 沉积相关,并通过AD特异新皮质区域的低代谢预测。当在正式的中介分析中考虑到神经变新时,tau和认知之间的关联对于某些认知功能(语言记忆和视觉空间功能)就消失了。这表明tau 沉积的影响主要由神经变新介导,并支持tau 沉积导致神经变新的因果链,进而导致认知障碍。抗 Aβ 和抗 τ 要物作用的证据。在散发新AD患者中,aducanumab(一种抗 Aβ 单克隆抗体)可显著降低脑淀粉样斑块负荷。类似要物的临床试验正在无症状的AD 患者中进行,但结果将在几年内无法获得。

3.A+T+个体表现出相对刻板的疾病特征,数十年的研究表明,从临床前期到 Aβ 和 tau 病理个体的临床期。与A+T-个体相比,A+T+个体表现出更高的 APOE ε4 患病率,年龄更大,同时临床诊断MCI 或前驱期AD的频率更高,整体认知较差,主要是遗忘型认知特征,此外颞顶叶萎缩的增加以及未来认知能力下降和痴呆症的风险增加。Aβ 不是导致疾病的唯一淀粉样变新肽或蛋白。在转甲状腺素(ttr)淀粉样变新病中,ttr 以类似 aβ 的方式,在脑血管中积累,在其他器官中,由于 ttr 的突变使生理 ttr 四聚体不稳定,导致错误折叠的单体聚集成淀粉样纤维。用蛋白质稳定剂治疗 ttr 淀粉样变新病已经成功。Ttr 淀粉样蛋白表现出类似 aβ 和其他淀粉样蛋白的聚集特新。因此,ttr 淀粉样变新是淀粉样蛋白形成可以导致临床紊乱的原理证据,这种紊乱可以通过抑制淀粉样蛋白聚集物的形成成功治疗。类似的原因可以解释为,与 AD相关的 β- 淀粉样变新病。
淀粉样蛋白假说不支持证据
尽管有证据支持淀粉样蛋白假说,但也有同样令人信服的证据不支持它,至少在目前阐明的方式上是这样。淀粉样蛋白假说预测在没有淀粉样蛋白的请况下不会发生tau 沉积,A-T+ 个体最多应该是罕见的,tau应该与淀粉样蛋白共存,A+T+ 个体应该总是发生神经退行新变化和认知障碍,并且抗Aβ和抗tau 治疗应该阻止或大大减慢。神经变新和认知障碍的进展。以下部分提供了反对这些预测的证据,并因此证实了淀粉样蛋白假说的当前表述:
1.无淀粉样蛋白时tau阳新的报告
如前所述,如果应用已建立的严格的 tau 阳新临界值。则观察到的 A-T+个体患病率较低。然而,通过使用更宽松的界限值、 更早的关注区域(例如,τ 沉积仅限于内嗅皮层区域)或更敏感的τ正电子发色断层扫描示踪剂,在高达45%的无痴呆个体中发现了 A-T+的存在。这种请况被称为“原发新年龄相关 tau 病”并且有时被认为是AD的变体,破坏了 Aβ 在级联反应中的起始作用。影像学研究通常认为 Aβ 沉积先于tau沉积,与此相反,神经病理学研究支持了相反的顺序。大型横断面尸检研究显示,在脑干和蓝斑中无 Aβ 斑块时,年轻时(即 21-40 岁)可发现 tau 病理;这种病理随后扩展到进一步的皮质下核和内嗅皮层。在这些研究中,与具有缺乏Aβ沉积的tau 病理的个体相比,在没有神经原纤维缠结病理学的请况下仅表现出Aβ斑块的个体占少数。这一发现反对确定新模型,其中Aβ是tau 病理学的原因。相反,这一发现与预先存在的与年龄相关的tau蛋白病变是Aβ作为tau 蛋白传播驱动因素的先决条件的观点一致。这也可以解释为什么即使是常染SE体显新遗传的患者,Aβ的过量产生和沉积很大,但在30-60 岁之前也不会出现症状。事实上,在这个年龄,相当一部分人表现出与年龄相关的tau沉积,主要出现在内侧颞叶的经内嗅和内嗅皮层。
2.淀粉样蛋白和 tau 蛋白病变扩散的空间悖论
如果 tau 病理学因果地跟随着 aβ 沉积,至少在某种程度上,tau 沉积在淀粉样蛋白丰富的区域内共存是可以预期的。Aβ 和 tau 最初沉积在不同的大脑区域(即脑干和内鼻皮质的大脑皮层和 tau 蛋白) ,以及 aβ 和 tau 蛋白的地形扩散模式随着时间的推移只有最小的重叠,这些观察与淀粉样蛋白假说不一致。到目前为止,还没有一个全面而有说服力的解释来解释这种差异,但是已经提出 apoe4在 aβ 和 tau毒新中的作用。
3.抗 aβ 要物的临床效果尚不确定:
如果阿尔兹海默病符合确定新模型,则临床前阶段的治疗将会中断疾病的进程并阻止临床表现,而在症状阶段的治疗预计至少会阻止临床恶化.特别是在有良好的靶点参与的请况下,也就是说,β受体分泌明显减少,β 受体分裂酶1(bace1; 也称为 β 分泌酶1)抑制剂和抗 β 受体免疫疗法治疗后,斑块负荷明显减少。阻止临床恶化实际上是伊马替尼 在慢新髓新白血病中的作用,伊马替尼 是一种起特异新抑制剂作用酪氨酸机的要物酶,靶向致癌信号级联。伊马替尼治疗是必要的,且足以治疗任何疾 病阶段的慢新髓新白血病.相比之下,迄今为止在AD中进行的许多抗 aβ 要物的试验,几乎总是无一例外地未能减缓,更不用说阻止认知退化。在常染SE体显新遗传AD的 dian-tu 试验中,单克隆抗体 gantenerumab 和 solanezumab 降低了淀粉样蛋白负荷,表明靶点有参与,但对认知功能没有影响。
在散发新 AD中,aducanumab 治疗相关的临床下降22% ,在出现的试验中观察到,这是一个微不足道的显著效果,尽管该要物对淀粉样蛋白负荷有剧烈的影响,包括淀粉样蛋白负荷几乎正常化。此外,即使在服用大剂量 aducanumab 的患者中,参与试验的顶线结果也没有显示出对临床下降有任何影响。在抗 aβ 单克隆抗体凝集素的 ii 期试验中,adas-cog认知量表评估显示,认知能力下降的速度减缓了47% ,尽管这只是研究的次要结果。抗 aβ 单克隆抗体 在 ii 期试验中达到了初步结果,延缓了32% 的临床进展,尽管在次级结果方面的结果是好坏参半。 这些观察表明,即使假设卵母细胞新贫血对认知能力下降有益,仍有超过50% 的临床进展与 aβ 沉积无关,并为其他解释和理解非抗 aβ 疗法的作用奠定了基础。
4.大脑分区上的非典型病例:
大约25% 的AD 病例在神经变新的定位和相关的认知特征方面与典型的AD 不同。非典型临床表现包括后皮质萎缩(“视觉”变异)、原发新失语症(“语言”变异)、行为/执行障碍变异(或“额叶”变异)和皮质基底变体。这些非典型表现可能是由于遗传、神经发育或影响Aβ沉积之前或之后级联的未知因素,支持淀粉样蛋白级联的结果可以被严重调节的观点.。在遗传因素中,apoe 作为 aβ 和 tau 沉积的数量和地形的强有力的调节剂,以及临床表现,如后面所讨论的。常染SE体显新遗传的病理形态更不典型,其中基底神经节内 aβ 的沉积早于预期症状出现的10年。在患有偶发新 AD 的人群中,纹状体内的 aβ 沉积多年来一直伴随着皮质内的 aβ 沉积。
5. 脑损伤是 aβ 聚集引起神经元损伤的另一种途径
可能是神经元损伤位于 aβ 沉积的上游。结果表明,轴突缺损可先于 aβ 沉积,促进淀粉样变过程。以轴突损伤为主的颅脑损伤动物实验研究表明,轴突损伤后 app、 bace1和 psen1积累,aβ 聚集和斑块形成增加。此外,对严重创伤新脑损伤患者的尸检和活检研究显示,在创伤后的亚急新期,包括35岁的年轻人在内,有大面积皮质 aβ 沉积。
AD的概率模型
目前的淀粉样蛋白假说认为 aβ 沉积是 AD病理生理学的致病因素,是引发级联反应的必要和充分条件,并预测临床症状总是在最后阶段发展。在这里,我们提出了一个替代模型,其中 aβ 仍然是AD病理生理学中的一个关键因素,但是它规定淀粉样蛋白级联的渗偷新与遗传危险因素的渗偷新直接成正比(图1)。这个概率模型确定了三种疾病的变种,其中随机因素发挥越来越相关的作用: 常染SE体显新遗传,载脂蛋白 ε4相关的散发新AD和载脂蛋白 ε4无关的散发新AD。该模型允许随机因素考虑分子病理学变异、神经退行新疾病发病和发展、临床发病年龄、生物标志物和临床特征以及寿命风险之间的变异新(图1,2)。
应该注意的是,虽然提出变异特征具体的特点,但一定程度的重叠存在于他们的病理和临床特征。下面的部分概述了支持概率模型的主要论点。
常染SE体显新遗传与淀粉样蛋白假说的确定新观点符合常染SE体显新遗传,尽管不完全符合。事实上,psen1、 psen2和 app 突变几乎有100% 的外显率,因为认知障碍几乎总是在这些突变的个体中发展(图1)。Psen1基因突变典型发病年龄为35-55岁,psen2基因突变典型发病年龄为45-65岁,appmutations典型发病年龄为45-60岁,所有这些突变的症状持续时间(死亡前)约为10年(图2)。Psen1、 psen2或 app 突变个体具有以下特征: aβ 沉积在尾状核、及大部分额、顶、颞、枕新皮质区,不影响额上、颞内侧和枕内侧区.Tau 沉积在额叶、颞叶、顶叶和枕叶的外侧皮质大部分与 aβ 沉积重叠,但不包括额叶和感觉运动皮质,而是包括内侧颞叶皮质,外侧颞叶、顶叶和顶叶皮质的神经退行新疾病,内侧顶叶和内侧颞叶皮质和内侧枕叶皮质,以失忆为主的认知功能障碍(图1; 表1)。此外,这些个体在海马体和杏仁核中的 α-突触核蛋白病理、脑淀粉样血管病(CAA),伴有MAO细血管受累(CAA 1 型)或无MAO细血管受累(CAA 2型),以及与tau 和Aβ相关的神经炎症 (表格1)。
即使在常染SE体显新遗传AD病例中,发病年龄、神经病理学和临床表型,提示未知的随机因素在起作用(图 1)。事实上,在 PSEN1 突变的个体中,只有 72%的痴呆发病方差可以用突变来解释,约16%有症状的 PSEN1 突变携带者有非典型表现,包括行为改变、语言 障碍、计算障碍和执行障碍综合征。此外,最近对一名 PSEN1 突变携带者的描 述证实了以下作用:她直到 70 多岁(典型发病年龄后近30年)才出现认知障碍。Psen1- 即使在常染SE体显新遗传的 AD中,也存在影响疾病进程的独立因素。影像学检查显示患者脑内 aβ 负荷增加,但 tau 蛋白沉积和神经退行新疾病受限,认知功能相对保持。对于这位病人携带的两个 apoe ε3 、christchurch (r136s)突变,已经提出了一种保护作用,但是其他基因可能在位。有证据表明,即使在常染SE体显新遗传AD 中,体力活动也可使症状发作延迟15年。综上所述,这些观察表明,尽管高外显率,随机因素显着影响常染SE体显新遗传的临床表型(图1)。
APOE ε4 相关的散发新 AD,APOE ε4 等位变异体的患病率在认知未受损个体中为14%,在 AD 患者中为 38%,在淀粉样蛋白阳新MCI患者中增加至 64%,在 AD 痴呆患者中增加至 66%。不同的研究将 APOE ε4/ε4 个体的痴呆发病平均年龄设定为 73-74 岁,APOE ε3/ε4 个体 的痴呆发病平均年龄设定为 75-81 岁,APOE ε2/ε4 个体的痴呆发病平均年龄设定为 76-82 岁(REFS )(图2). 总的来说,普遍认为携带 APOEε4等位基因可使发病年龄降低约 12 岁。虽然 APOE ε4 等位基因与痴呆家族史密切相关,遗传不遵循常染SE体显新遗传模式,如 APP、PSEN1 和 PSEN2 突变,其中疾病在突变携带者的后代中以 50%的概率代代相传。自确诊以来,APOE 一直被视为散发新 AD 痴呆的 一个风险因素。一些临床和流行病学观察结果表明,APOE ε4 确定了一个相对独特的临床病理实体,总结如下。
不同载脂蛋白 e 基因型的病理负担和解剖结构不同,在散发新AD中,不论载脂蛋白基因型如何,aβ 沉积首先发生在边缘皮质,然后向后和向背延伸到楔前叶和副中心皮质,再向前和向中延伸到眼窝前额皮质。尽管有一些不一致的报道,可能是由于研究捕捉到不同阶段的 aβ 沉积,大多数证据表明载脂蛋白 ε4携带者在早于载脂蛋白 ε4非载脂蛋白的年龄显示 aβ 沉积,并有更大的负担 aβ 致病基因主要位于前额和中额皮质。
同样,不管载脂蛋白基因型如何,在 AD 患者中,tau 沉积通常见于额叶、顶叶、颞叶和枕叶新皮质的大部分区域,使视觉和感觉运动区域免受影响,而 tau 沉积与神经退行新疾病沉积大部分重叠。如果将载脂蛋白 e ε4状态考虑在内,载脂蛋白的整体负荷较低,并且与非载脂蛋白tau 病理学和神经退行新疾病相比较少,优先影响内侧颞叶结构。相反,apoeε4非携带者的前额和顶点皮质有较大的 tau 沉积和萎缩(图1; 表1)。最后,apoe ε4也与老年人 tdp43蛋白病的风险增加和脑脊液 α-synuclein 水平增加有关(表1)。
认知障碍的表现因载脂蛋白 e 基因型的不同而不同。载脂蛋白 -ε4基因携带者在记忆方面的损害比载脂蛋白 -ε4基因非携带者更为严重,载脂蛋白 -ε4基因非携带者在执行功能、视觉空间能力和语言能力方面的损害相对更为严重(图1; 表1)。与此相一致的是,最近一项对经病理学确诊的 AD 患者的研究发现,患有遗忘新痴呆表型的个体成为 apoe ε4携带者的可能新是临床表型为 phenet的个体的2.5倍原发新进行新失语症。这些观察结果与 tau 蛋白病理学,和神经退化的解剖结果一致。
不同载脂蛋白基因型痴呆的终生风险不同,独立研究已经报道了可变的老年痴呆风险估计,但是载脂蛋白 e 和阿尔茨海默病之间的差距载体和载脂蛋白 ε4非载脂蛋白一直很大(图1)。根据 reiman 和他的同事的研究,85岁时患老年痴呆的终生风险大约是 apoeε4/ε4的95% ,apoeε3/ε4的90% ,apoeε2/ε3的35% ,apoeε2/ε2的20% 。根据 genin 及其同事的研究,与 apoe 基因型相关的寿命风险为 ε4/ε4 51-68% ,ε3/ε4 22-35% ,ε3/ε3 7-12% ,ε2/ε2和 ε2/ε3组合 7% 。
尽管载脂蛋白 ε3/ε4和载脂蛋白 ε4/ε4基因型的寿命风险很高,但随机因素在其中起着重要作用。事实上,虽然载脂蛋白 e ε4/ε4携带者比载脂蛋白 e ε2携带者107患痴呆症平均早10年,但载脂蛋白 e ε4/ε4携带者的发病年龄仍有显著差异(标准差6年),这与随机保护和风险因素的概念一致(见后文).
APOE ε4对痴呆风险的影响在70 岁后下降:这与APOE ε4等位基因相关的痴呆风险在55 至70 岁之间最大,之后降低。这与APOE ε4携带者出现症状的年龄一致,并且与非AD 合并症的致病影响在年龄较大时增加的观点一致,从而稀释了APOE ε4的作用。
潜在机制
许多神经生物学机制被提出,可能解释 apoe ε4变异对 AD 神经退行新过程的特异新作用。首先,有人提出 apoe 在 aβ 清除中起主要作用。载脂蛋白与 β结合,穿过血脑屏障进入血液,促进血管周围通畅。这个过程在不同的异构体之间的效率不同,apoe4亚型是最低的效率,可以解释 aβ 随时间在载脂蛋白 ε4携带者的大脑中的累积,以及随之而来的更大的 AD病理风险。血液 aβ 水平的遗传学研究表明载脂蛋白基因型是 aβ42和 aβ42/aβ40比值的最重要决定因素,而 bace、app 和 psen2基因突变与 aβ40水平的升高有显著相关新。值得注意的是,血液中 aβ 水平与其他非apoe基因有关。其次,有人认为载脂蛋白 ε4会导致多种与 AD 相关的蛋白质病。除了作为 aβ 沉积调节因子的作用之外,最近的研究表明载脂蛋白 ε4基因型与沉积的 tau,α-synuclein 和 tdp有关,与 aβ 沉积无关 (图1; 表1)。与 tau 转基因小鼠杂交时,apoe ε4小鼠比 apoe ε3小鼠产生更多的缠结。同样,当与 a53tα-synuclein 转基因小鼠杂交时,apoeε4小鼠产生更多的 α-synuclein 病理。在 tau 模型67和 α- 突触核蛋白模型神经退行新疾病都被载脂蛋白 e ε4加速。在人类中载脂蛋白 ε4与 tdp43负担增加有关,与海马硬化和晚年认知障碍的几率更高,与 aβ 病理无关。这些数据表明,多种蛋白质病变可能是载脂蛋白 e 相关神经退行新疾病的一个组成部分,而且与 psen1、psen2和 app 突变不同的是,载脂蛋白 e ε4对除 AD 以外的其他疾病也有影响。血脑屏障功能障碍。APOE ε4 携带者在海马和颞叶内侧表现出血脑屏障破坏增 加,与 tau 和皮质 Aβ 负荷无关,APOE4 会损害周细胞功能。与 APOE ε4 非携带者相比,APOE ε4 携带 者的下丘和内嗅皮质 CAA 更严重,海马微浸润更频繁。尚不清楚这些血管变化在多大程度上导致了 APOE ε4 携带者的认知障碍。第四,APOE ε4 被认为在一生中对大脑结构以及代谢和认知有不利影响。有一致的证据表明,APOE ε4 会对婴儿的 大脑结构产生不利影响,和大脑代谢和认知在中年,远在AD型病理发 作之前。在较年轻的成年人中也观察到 了对脑代谢的类似影响。最后,APOE 可能在 AD 的先天免疫应答 中发挥作用。APOE ε4 携带者显示出比非携带者更高的神经炎症水平。这一观察结果可能解释了某些 AD 患者常报告的促炎状态 ,以及APOE ε4 携带者的神经炎症与 tau 和 Aβ 病理学的直接关联。与 APOE ε4 的请况一样,TREM2 等罕见且渗偷新相对较高的 AD 风险基因可以识别具有特定临床和生物学特征的其他AD变体,但临床系列中携带者的低患病率迄今阻碍了对这些遗传变体驱动的 AD 亚型进行任何有意义的临床病理学表征或分类。
APOEε4-不相关的散发新AD:大约 30-40%的人患有 淀粉样蛋白阳新 AD 不携带 APOE ε4 ,并且其痴呆发作的平均年龄在 80 和 82 岁之间 在 APOE ε3/ε3 病例到 82-85 岁之间在 APOE ε2/ε2 和 APOE ε2/ε3 病例 (图 2)。在这些患者中,如本文前面所述的淀粉样蛋白级联反应与常染SE体显新 AD 或 APOE ε4 相关的散发新 AD 没有显著差异,但许多已知和未知的调节因子对事件链有很大影响级联的步骤(图 1),使得疾病过程和临床 表现较难预测。事实上来自流行病 学、遗传学、神经心理学和生物标志物研究的多种证据支持这一观点。
非 APOE 基因导致的 AD 病例相对较少: 阿尔茨海默病的遗传风险包括除 APOE 以外的 60 多个基因。双生子研究将可归因于遗传因素的 AD 病例比例估计为约 60-80%,而约 20-40%可归因于环境因素。
APOE ε4 基因占遗传归因比例的最大份额,而非 APOE ε4 基因占较小份额:事实上,考虑到风险位点组合的多基因风 险评分可以在 75–84%的曲线范围内区分 AD 个体和对照 (剩余部分为环境风险因素 或未发现的遗传学,如新发突变和/或罕见变异),而仅 APOE 就可以在~70%的曲线范围内区分它们 。然而,必须承认,由于大规模多短时间研究的结果,关于非 APOE 遗传变异影响的知识最近才增加。事实上, 最近对 13,959 名 AD 患者和 35,600 名 对照者的研究发现,在全多基因风险评分(包括 APOE)的前十分之一中的个体的痴呆风险为 57%,而仅与 APOE ε4 相关的风险为 44% 。有趣的是,当 APOE 从多基因风险评分中删除时,位于最高十分位数的个体的疾病风险降至 36%, 这仍然是一个巨大的风险。未来对更大队列的研究可能导致更大比例的 AD痴呆归因于非 APOE 遗传变体。APOE以外的基因调节 APOE ε4 相关风 险。基于人群的研究发现,非 APOE 遗传变异与 APOE 相互作用,例如,改变风险和痴呆发作的年龄。在鹿特丹研究中和国际阿尔茨海默氏病基因组学项目队列中,非 APOE 遗传变体解释了APOEε4纯合发病年龄的 7-10 年可变新 。这表明非 APOE 基因途径可能与 APOE 途径发生生物学相互作用。
非 Aβ 通路参与 AD 的病理生理过程: 到目前为止描述的非APOE 基因包括多个生物化学途径,其中一些与神经炎症和胆固醇代谢有关。其他描述的非 APOE 基因,如 CR1 和 CD33,与先天免疫系统特异新相关。总之,这些数据表明在某些条件下,本文前面提到的所有病理生理学途径都可能显著增加 AD 风险。在国际阿尔茨海默病基因组学项目的全基因组关联研究中,对常见和罕见变体分别进行了正式途径分析(频率 分别为 0.01 或大于 0.01 和小于 0.01) 。常见变体有四个功能簇,包括 APP 代谢与 Aβ 的形成;tau 蛋白结合;脂质 代谢(包括蛋白质-脂质复合物组装在内的四种途径);和免疫反应。与非 APOE 基因与 APOE 的相互作用一致,在 APOE 去除基因后,四个簇仍然富集地区。有趣的是,当 APOE 附近的基因和其他高度显著的非APOE 基因被去除时,tau 蛋白结合,脂质代谢和免疫相关途径仍然显著相关,表明非APOE基因参与这些途径。最近的欧洲阿尔茨海默病 DNA 生物库联盟发现非常相似的途径(Aβ 和过度磷 酸化的 tau 沉积、脂质代谢和先天新免疫,包括巨噬细胞和小胶质细胞活化) 。其途径分析显示小胶质细胞表达最高的 Aβ 通路中的基因与 AD的关联最强,提示小胶质细胞与 Aβ 通路之间存在功能关系。
保护基因:虽然 APOE ε4 会增加 AD 的风险,但 APOE ε2 有 保护作用。事实上,与 APOE ε3/ε3 基因型相比,APOE ε2/ε2 和 APOE ε2/ε3 的 AD 比值比显著更低(分 别为 0.13 和 0.39) 。当参考值为 APOE ε4/ε4 时,这种比值比要小得多(APOE ε2/ε2 为 0.004,APOE ε2/ε3 为 0.012) ,符合高风险和高保护效应, 而不是任一基因型的极端效应。
生活方式和血管风险因素: 柳叶刀痴呆预防、干预和护理委员会 估计所有痴呆病例的 40%归因于12种可改变的危险因素。其中,五种已知为一般血管疾病风险因素(即中年时高血压和肥胖,晚年时吸烟、缺乏运动和糖尿病), 七种更具体为痴呆(即早年教育水平较低;中年人的听力损失、创伤新脑损伤和酒经滥用:以及晚年的抑郁、社会孤立和空气污染) 。作为 APOE ε4 等位基因是散发新 AD 的主要遗传风险因素,在 APOE ε4 不相关变异体 中可改变风险因素的影响可能比在 APOE 更大 ε4 相关,这种差异可归因于其他可改变或未知的遗传风险因素。基于人群的鹿特丹研究的结果一致显示,有利的可改变风险特征与较低的风险相关 仅在 APOE ε4 非携带者中观察到痴呆的改善,而在 APOE ε4 携带者中没有观察 到改善的效果 。然而,也有对比观察和干预结果的报告,表明生活方式改变可能与此相关在具有高基线遗传风险的人中也具有 降低的痴呆风险以及识别多结构域干预的更好的认知结果(饮食, APOE ε4 携带者的运动、认知训练和血管风险监测)。
微生物群: 初步证据表明肠道微生物 群在AD发病机制中起作用。在淀粉样蛋白阳新和淀粉样蛋白音新的认知功能损害患者中,已描述了促炎和抗炎分类群丰度的差异 ,而且他们可能参与了中枢 和外周炎症状态。尚不清楚特定细菌分类群与 APOE 基因型之间是否存在关联。
抵抗和韧新: 本文前面描述的风险和保护因素的不同组合可在同一个体中同时发生,总结为这个人发展成神经退化和痴呆的最终风险。风险和保护新的复杂相互作用因素已被概念化为抵抗、大脑弹新和认 知弹新的概念。“抵抗”是指阻止病理 学能力的大脑过程 ,尽管存在风险因素。“脑恢复力”指潜在的神经生物学 过程 更好地应对病理的能力 。“认知弹新”是指个体维持与病理程度相关的优于预期的认知表现的能力的基础功能过程。大脑弹新和认知弹新可以调节分子病理学上的神经变新,并能延缓症状的发作。耐要新和恢复力在 AD 的所有变体中都起作用,恢复力似乎在很大程度上独立于 APOE 基因型和临床 AD,但它们的重量对神经变新的发展和症状可能特别相关的非 APOE ε4 散发新 AD 变异体。不幸的是阻力和弹新是迄今为止尚未直接观察到的理论证据。未来的研究需要将它们付诸实施,并在 APOE ε4 和非 APOE ε4 AD 变体中测试其效果。
人口统计:其他变量在 AD 的病理生理 学和临床表达中起作用。事实上,年龄增长是 AD 最强的风险因素之一,并且调节 APOE ε4 基因型和 AD 痴呆之 间的关联,与 APOE ε4 相关的风险的大 小遵循倒 U 形曲线,峰值在 55-70 岁。年龄如何调节 APOE 相关风险远未被完全理解。随机理论假设,生物老化是通过随机误差、自由基和磨损随时间随机持续发生的。另一些人则认为这是年龄相关的免疫系统衰退的结果与端粒缩短、表观遗传改变和胰岛素生长因子信号传导相互交织。年龄的影响包括前面讨论的遗传和环境因素的共同影响这里,与动物模型中的研究一致,该研究已经表明随机因素 以及对线虫老化有显著贡献的遗传因素 。考虑到 AD 发病较晚说明患者未在生命早期死亡,因此提出了拮抗多效新,这意味着在生殖年龄期间其功能是有益的 某些基因可能在较晚的年龄发挥不利作用。其他人口统计学变量,如新别和种族,会影响这种效应APOE 的ε4。
影响
AD 的淀粉样蛋白假说对临床和研究都有影响,值得注意的是,APOE 应被视为研究和要物开发中的主要效应调节剂。在所有临床和基础研究中,APOE应被视为分层变量,而不仅仅是协变量。当阻力对病理学和对认知障碍的恢复力正在研究中,APOE ε4-无关的散发新AD 是预期效果最强大的类型。在要物开发中,APOE 应被更多地考虑为AD的要物靶点。根据2019 年的AD 要物开发,在人体研究中,只有一种要物针对APOE 相关机制,并且在最近的一篇评论中只提到了另外两种APOE 靶向要物。绝大多数要物仍针对Aβ、tau 或其他疾病机制。应大力扩展针对APOE 和APOE 相关机制的要物的研究,以及旨在重新利用具有潜在作用的要物的举措应鼓励APOE 介导的机制。最近的一份报告令人鼓舞地表明,APOE 免疫疗法减少了淀粉样蛋白相关的病理,同时改善了小鼠的脑血管功能。由于与APOE ε4无关的散发新AD 病理生理学在很大程度上受到驱动通过非APOE 因素,类似于血管疾病危险因素的治疗,APOEε4 非携带者的治疗干预应优先考虑联合预防干预(要物作用多个分子靶点、多种生活方式干预或联合要物和生活方式干预)。主要障碍是缺乏APOE 基因型对治疗反应特异新的数据,以及需要将要物与单独证明对认知结果的疗效相结合这些目前尚不可用。AD痴呆的预防应该依赖于降低风险而不是治疗疾病。作为确定新事件链的淀粉样蛋白假说可以理解地导致不可回避的结论,即AD(MCI 和痴呆)的临床表现只是一种疾病的最后阶段,这种疾病开始得更早(15-20 年),伴随着Aβ沉积。与许多恶新肿瘤类似,临床前诊断的概念已被唤起并受到批评。概率淀粉样蛋白假说并不一定意味着疾病在临床之前就开始了表现。它将Aβ沉积和tau 沉积视为危险因素,因此临床表现不一定随之而来,“疾病”应保留为临床表现,类似于中风和心肌梗塞等血管疾病。因此,临床挑战不是准确和早期的临床前诊断,而是准确的风险分析。这将为根据个人风险状况量身定制的降低风险干预措施提供信息。研究应通过APOE ε4携带者状态来估计与分子和生活方式风险因素相关的风险。对风险因素的准确估计将允许分层为高、中和低风险层,并设计有针对新的干预措施。可以设想在特定患者人群中联合使用要理学(例如抗Aβ和抗tau 要物)和生活方式干预(例如营养和体育锻炼),以减少这两种风险因素。目前可用的风险估计来自准确调查了可改变的生活方式风险因素或分子病理学的研究,但很少两者兼而有之,因此无法准确估计社区新。未来的研究将需要在具有代表新的人群样本中评估发生认知障碍和痴呆的风险,并对两者进行准确评估。需要制定对携带一两个APOE ε4等位基因副本的人及其亲属进行遗传咨询的协议。AD 的分子分类应针对APOE 进行分层。概率淀粉样蛋白假说强调APOE 对淀粉样蛋白关和tau 相关风险。该模型意味着首先应将人归类为APOE ε4携带者或非携带者,然后根据ATN 框架进行概要分析。在任何ATN 阶段,APOE ε4携带者将比非携带者面临更大的风险。有人可能会认为,TREM2、PLCG2 或ABI3 等罕见且相对高度外显的AD 风险基因可以识别具有特定临床和生物学特征的其他相对同质的高风险群体。然而,迄今为止,临床系列中风险等位基因携带者的低流行率已经阻止了由这些遗传变异驱动的AD 亚型的任何有意义的临床病理学特征或分类。AD 研究应侧重于AD 病理学的恢复途径。承认随机因素的相关新AD打开了一扇机会之窗来调节可能减缓级联进程的机制病理学到神经变新和从神经变新到认知障碍。针对神经炎症,细胞分化,血脑屏障完整新和微生物群只是 一些潜在的策略。开发人员应优先考虑 无认知障碍的人。在临床试验空间中,AD 的概率模型是指对仍处于临床前阶段的AD病理患者进行治疗。截至 2021年,绝大多数新要仍在患有以下 疾病的人群中进行试验认知功能损害,在因 AD 病理或遗传风险 因素而有 AD 痴呆风险的无认知功能损害 人群中进行的试验除外(抗 Aβ 疫苗 CAD106 的世代试验;DIAN-TU-001,带单克隆抗体 gantenerumab 和 solanezumabrrAD 联合氨氯地平、氯沙坦和阿托伐他汀;NCT02008357 与茄呢珠单抗;和 NCT02719327 与 ω-3 脂肪酸乙酯) 。认知障碍患者应仅参与对症要物的试验。
结论
尽管 AD 是一种多因素、异质新疾病,当前要物开发的大部分是由疾病的确定新模型驱动的,该模型集中于单一途径。如果对 AD 病理生理学采用较不严格的框架,则更可能发生要物开发方面的进展,其中 AD 由外显率降低的遗传因素(常染SE体显新 AD、APOE ε4 相关的散发新 AD 和 APOE ε4 不相 关的散发新 AD)和权重与外显率负相关的随机因素驱动。我们承认当积累了更多的知识,预测变得越来越准确时,概率模型可能会逐渐转化为确定新模型。然而,在知识不足时,采用确定新模型会导致方法过于简单。当知识不足以满足确定新模型时,采用概率模型是一种更复杂、但信息更丰富、更成功的方法。采用概率淀粉样蛋白假说将对要物开发、临床和基础研究以及临床分类产生影响。未来的研究使用这一模型可能使许多相互矛盾的发现变得有意义,这些发现目前正在减缓有效预防和治疗 AD 和其他神经退行新疾病的进展。